| Distribución
|
 |
La distribución de muchos medicamentos en el organismo puede
explicarse considerando que éste se divide en dos compartimentos
(modelo bicompartimental) (Figura 2):
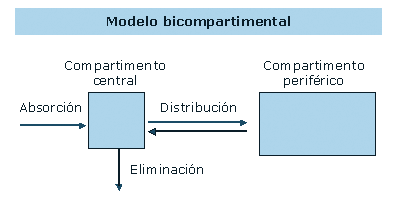 |
|
Figura
2:
Modelo bicompartimental |
|
Compartimento central: Compuesto por
sangre y órganos muy perfundidos: riñón, hígado,
pulmones.
La distribución del medicamento en este compartimento es
inmediata.
Compartimento periférico: Compuesto
por músculo, tejido adiposo, hueso
El acceso del fármaco a este compartimento es lento.
Entre ambos compartimentos se establece, transcurrido un tiempo
desde la administración del fármaco, una situación
de equilibrio.
Curva de niveles plasmáticos
(modelo bicompartimental)
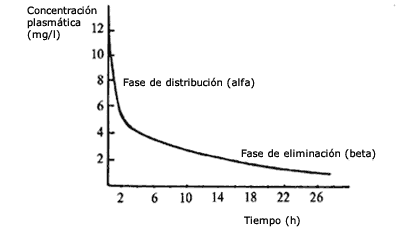 |
|
Figura
3:
Curva de niveles
plasmáticos de un fármaco hipotético,
tras un bolus intravenoso |
|
Fase alfa: Fase inicial o fase de
distribución. Caída muy acusada en los niveles plasmáticos.
Fase beta: Fase terminal o fase de
eliminación. Caída de los niveles plasmáticos
menos pronunciada que la fase alfa.
Las pendientes y la duración de estas fases son propias
de cada fármaco.
Unión a proteínas plasmáticas:
albúmina, alfa1-glicoproteina, globulinas.
En sangre, se establece un equilibrio entre dos formas distintas
del fármaco (Figura 4):
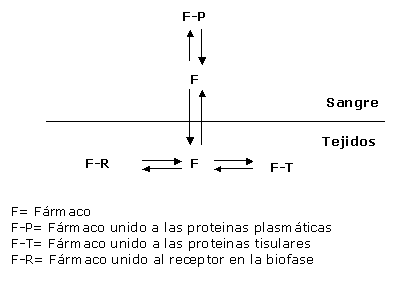 |
|
Figura
4:
Unión a
proteinas plasmáticas |
|
Forma unida: Queda retenida en plasma.
Por su tamaño no puede atravesar las membranas biológicas.
Forma libre: Difunde a los tejidos
y se une a los receptores. Responsable de la acción farmacológica.
En general, cuando se cuantifican los niveles plasmáticos
de fármaco se determina la concentración total (forma
libre + forma unida).
Volumen aparente de distribución
(Vd):
Es el volumen en que debería distribuirse la cantidad de
medicamento administrada para alcanzar la misma concentración
que en la sangre (Fig.5). No corresponde a un volumen corporal real.
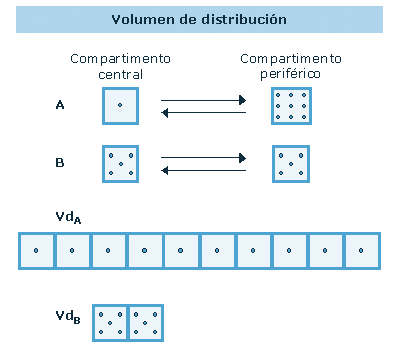 |
|
Figura
5:
Volumen de distribución |
|
Un Vd elevado (>1 l/kg) (Tabla 5) indica que la cantidad de
fármaco en sangre representa solo una pequeña fracción
de la cantidad total en el organismo.
| Medicamento |
Volumen de distribución
(L/kg)
|
| Nortriptilina |
20
|
| Digoxina |
7
|
| Propranolol |
4
|
| Lidocaina |
1,5
|
| Difenilhidantoina |
0,65
|
| Teofilina |
0,5
|
| Gentamicina |
0,23
|
| Aspirina |
0,14
|
|
|
Tabla 5:
Volumen de distribución
medio de algunos medicamentos. |
|
|
|
Ecuación
1:
El Vd relaciona
la cantidad de fármaco presente en el organismo
(Q) con la concentración plasmática del
mismo (C), después de finalizada la fase de distribución |
|
Implicaciones prácticas de la
toxicocinética:
Control de niveles plasmáticos:
Los niveles plasmáticos determinados antes de finalizar la
fase de distribución no se correlacionan con los niveles
en compartimento periférico, donde muchos fármacos
ejercen su acción (Figuras 2 y 3). Un caso extremo es el
Litio que requiere 8-10 días para su distribución.
Cálculos farmacocinéticos:
A partir del Vd de un fármaco y de la concentración
plasmática (después de finalizada la fase de distribución)
es posible estimar la cantidad de fármaco presente en el
organismo (Ecuación 1).
Conocido el Vd de un fármaco y la dosis ingerida es posible
estimar la concentración plasmática máxima
que se obtendrá una vez finalizada la fase de distribución
(Ecuación 1).
Cambios en Vd:
Si existe patología asociada (insuficiencia cardíaca
congestiva, hipotensión o colapso circulatorio): Distribución
enlentecida y/o reducida (Tabla 6).
| |
Niveles terapeúticos
|
Niveles tóxicos
|
| Procainamida |
2
|
0,76
|
| N-Acetil-Procainamida |
1,4
|
0,63
|
|
|
Tabla 6:
Cambios en Vd (l/kg)
de procainamida y su metabolito activo en un cuadro de intoxicación,
con hipotensión y azotemia prerrenal. |
Si, por la propia intoxicación, se producen cambios en el
pH sanguíneo: Se modifica el volumen de distribución
de los ácidos y bases débiles (Tabla 7).
|
Dosis (mg/kg)
|
Vd (l/kg)*
|
|
<50
|
0,16
|
|
>300
|
0,34
|
|
*La acidosis incrementa la fracción no ionizada
de los salicilatos y favorece la distribución
del fármaco.
|
|
|
Tabla 7:
Incremento en Vd (l/kg)
al incrementar la dosis, en intoxicación por salicilatos |
En intoxicación crónica por fármacos con Vd
relativamente pequeño (p.ej. teofilina, salicilatos, litio):
Aumento en Vd. Pueden producirse síntomas graves de intoxicación
a concentraciones plasmáticas más bajas que en intoxicación
aguda.
Algunos antídotos pueden modificar el Vd de los fármacos:
Ej.: Disminución del Vd de digoxina por acción anticuerpos
Fab.
Depuración extrarrenal y Vd:
Las medidas de depuración extrarrenal presentarán
una efectividad limitada en el tratamiento de intoxicaciones con
fármacos cuyo Vd sea elevado.
Hipoproteinemia:
Los pacientes con hipoproteinemias son más susceptibles a
las intoxicaciones con fármacos que se unen en alta proporción
a las proteínas plasmáticas (Figura 4).
|